Parametrization of metallo-proteins always been a challenging problem during the set-up of classical molecular dynamics simulation. This tutorial will describe a short-cut way to set up and Zinc (Zn2+) co-ordinated metallo-protein where Zinc stays in co-ordination with two histidines (HID) and one glutamate as described in the following figure (Figure 1). Note: All the waters are explicitly treated.
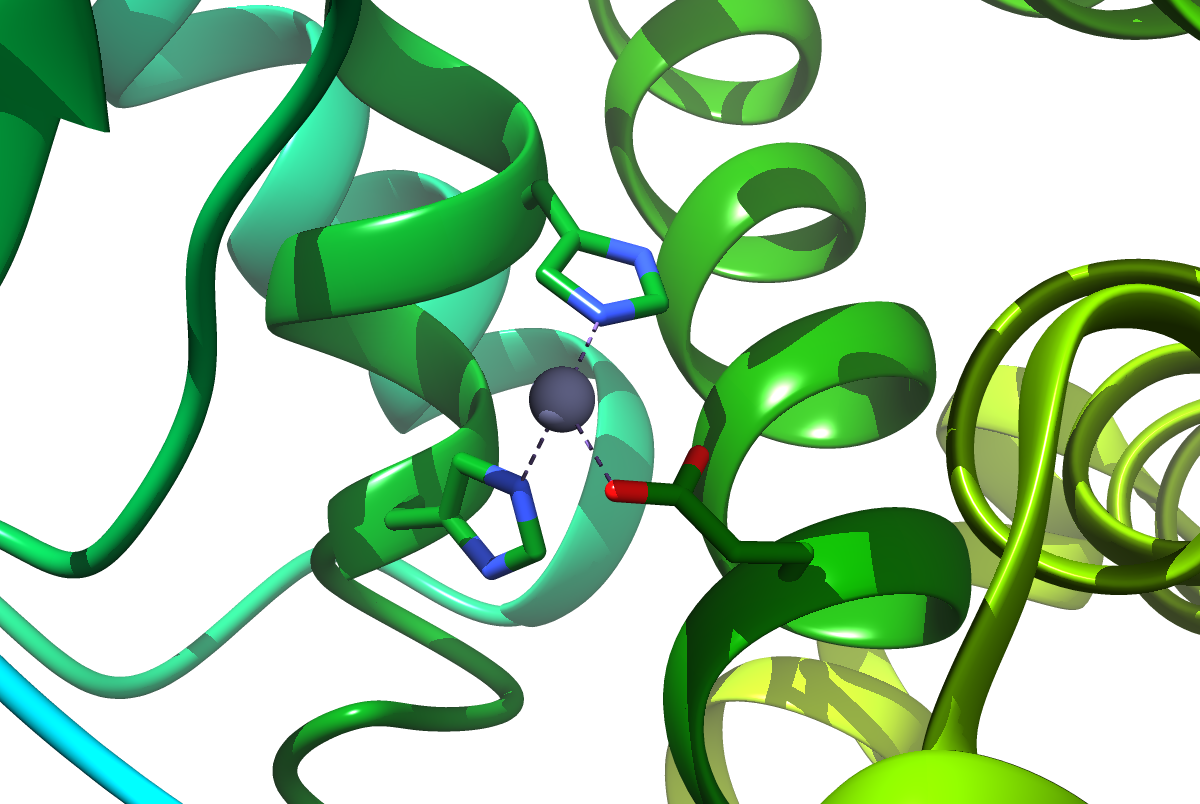
Figure 1. Zn co-ordinated with two histidines and one glutamate.
The parameters for this Zn and active site residues can be found here. The work has been carried out by Yang et al. See their paper here.
Step by step guide on how to execute this:
Pre-requisites:
- tleap
- Acpype to convert Amber topology and coordinates to Gromacs compatible versions.
- H++ server.
- Downloaded files in a particular folder
1. Upload your PDB in H++ server and set the protonation state (for example pH 7.0).
2. Download the output PDB (Make sure you modeled all non-terminal missing residues using Modeler or using any other packages).
3. Now rename two histidines that are bound to ZINC as HID instead of HIS and visualize the structure to see if this looks logical or not?
4. Inside M1_chg2.prep file you will see the histidines are named as HD1 and HD2 whereas glutamate is named as GL1. Edit the names in your PDB files. See below.
ATOM 2392 N HD1 301 -18.445 5.436 9.635 1.00 0.00 N ATOM 2393 CA HD1 301 -18.898 4.830 10.883 1.00 0.00 C ATOM 2394 CB HD1 301 -20.360 4.381 10.785 1.00 0.00 C ATOM 2395 CG HD1 301 -20.924 3.935 12.100 1.00 0.00 C ATOM 2396 ND1 HD1 301 -21.549 4.797 12.971 1.00 0.00 N ATOM 2397 CE1 HD1 301 -21.908 4.137 14.063 1.00 0.00 C ATOM 2398 NE2 HD1 301 -21.523 2.880 13.931 1.00 0.00 N ATOM 2399 CD2 HD1 301 -20.904 2.727 12.714 1.00 0.00 C ATOM 2400 C HD1 301 -18.018 3.660 11.319 1.00 0.00 C ATOM 2401 O HD1 301 -17.449 3.688 12.388 1.00 0.00 O ATOM 2424 N HD2 305 -15.025 3.292 14.546 1.00 0.00 N ATOM 2425 CA HD2 305 -14.974 2.206 15.529 1.00 0.00 C ATOM 2426 CB HD2 305 -15.786 0.998 15.064 1.00 0.00 C ATOM 2427 CG HD2 305 -17.207 1.024 15.511 1.00 0.00 C ATOM 2428 ND1 HD2 305 -17.561 1.168 16.834 1.00 0.00 N ATOM 2429 CE1 HD2 305 -18.884 1.160 16.932 1.00 0.00 C ATOM 2430 NE2 HD2 305 -19.390 0.999 15.723 1.00 0.00 N ATOM 2431 CD2 HD2 305 -18.367 0.901 14.814 1.00 0.00 C ATOM 2432 C HD2 305 -13.542 1.760 15.815 1.00 0.00 C ATOM 2433 O HD2 305 -13.223 1.284 16.920 1.00 0.00 O ATOM 2587 N GL1 324 -21.029 3.090 20.760 1.00 0.00 N ATOM 2588 CA GL1 324 -21.816 3.640 19.651 1.00 0.00 C ATOM 2589 CB GL1 324 -23.171 2.938 19.536 1.00 0.00 C ATOM 2590 CG GL1 324 -23.080 1.441 19.199 1.00 0.00 C ATOM 2591 CD GL1 324 -22.408 1.155 17.857 1.00 0.00 C ATOM 2592 OE1 GL1 324 -22.275 2.081 17.022 1.00 0.00 O ATOM 2593 OE2 GL1 324 -22.020 -0.009 17.626 1.00 0.00 O ATOM 2594 C GL1 324 -22.002 5.163 19.683 1.00 0.00 C ATOM 2595 O GL1 324 -22.066 5.793 18.636 1.00 0.00 O
5. Rename the rest of histidines as either HIP, HIE or HID based on the following
HID: Histidine with hydrogen on the delta nitrogen
HIE: Histidine with hydrogen on the epsilon nitrogen
HIP: Histidine with hydrogens on both nitrogens; this is positively charged.
6. Remove all the hydrogens from the amino acids.
7. Make sure the Zn is separated from the amino acids using a TER. Something like following. Renamed the ZN as ZN1 as in M1_chg2.prep file it is named as ZN1. Rest of the ZINC should be ZN not Zn.
ATOM 5146 CB LEU 632 -45.178 -16.254 12.289 1.00 0.00 C ATOM 5147 CG LEU 632 -45.901 -16.066 10.953 1.00 0.00 C ATOM 5148 CD1 LEU 632 -44.934 -16.199 9.786 1.00 0.00 C ATOM 5149 CD2 LEU 632 -47.046 -17.057 10.822 1.00 0.00 C ATOM 5150 C LEU 632 -45.155 -16.312 14.792 1.00 0.00 C ATOM 5151 O LEU 632 -44.462 -15.489 15.391 1.00 0.00 O ATOM 5152 OXT LEU 632 -44.797 -17.516 14.897 1.00 0.00 O TER ATOM 1 ZN ZN1 633 -21.374 1.098 15.223 1.00 0.00 ZN END
8. Run the tleap. See the tleap input below
source leaprc.ff14SB loadamberparams frcmod.ionsjc_tip3p loadamberprep ~/Dropbox/LTA4H_collab/M1_chg2.prep loadamberparams ~/Dropbox/LTA4H_collab/M1.frcmod loadamberparams ~/Dropbox/LTA4H_collab/metals.frcmod x = loadPDB test.pdb savepdb x pdbout bond x.301.NE2 x.633.ZN bond x.305.NE2 x.633.ZN bond x.324.OE1 x.633.ZN check x saveAmberParm x prot.top prot.crd charge x addIons2 x Na+ 0 solvateOct x TIP3PBOX 10.0 saveAmberParm x prot_solvated.top prot_solvated.crd savepdb x prot_solvated.pdb quit
Use the following command to run tleap
tleap -s -f tleap.all
If you get any error please check the leap.log and post it as comments.
9. You will get AMBER generated topology and coordinates as outputs.
10. Use acpype to convert them to Gromacs compatible version. See here for the usage of acpype.

3 responses to “Parameter for zinc centre proteins”
Can we also use this method for generating parameters of protein contains Iron (Fe) metal.?
LikeLike
Wooo..no idea however I think Autodock has parameters for all the metalloprotease. Check here http://autodock.scripps.edu/faqs-help/faq/faqsection_view?section=Scientific%20Questions Unfortunately I don’t have a first hand experience with Fe docking. But the method could be similar.
LikeLike
I am not asking about Iron (Fe) parameter for docking, I want to generate the topology and coordinate files for MD simulation of a system that have iron as a metal.
LikeLike