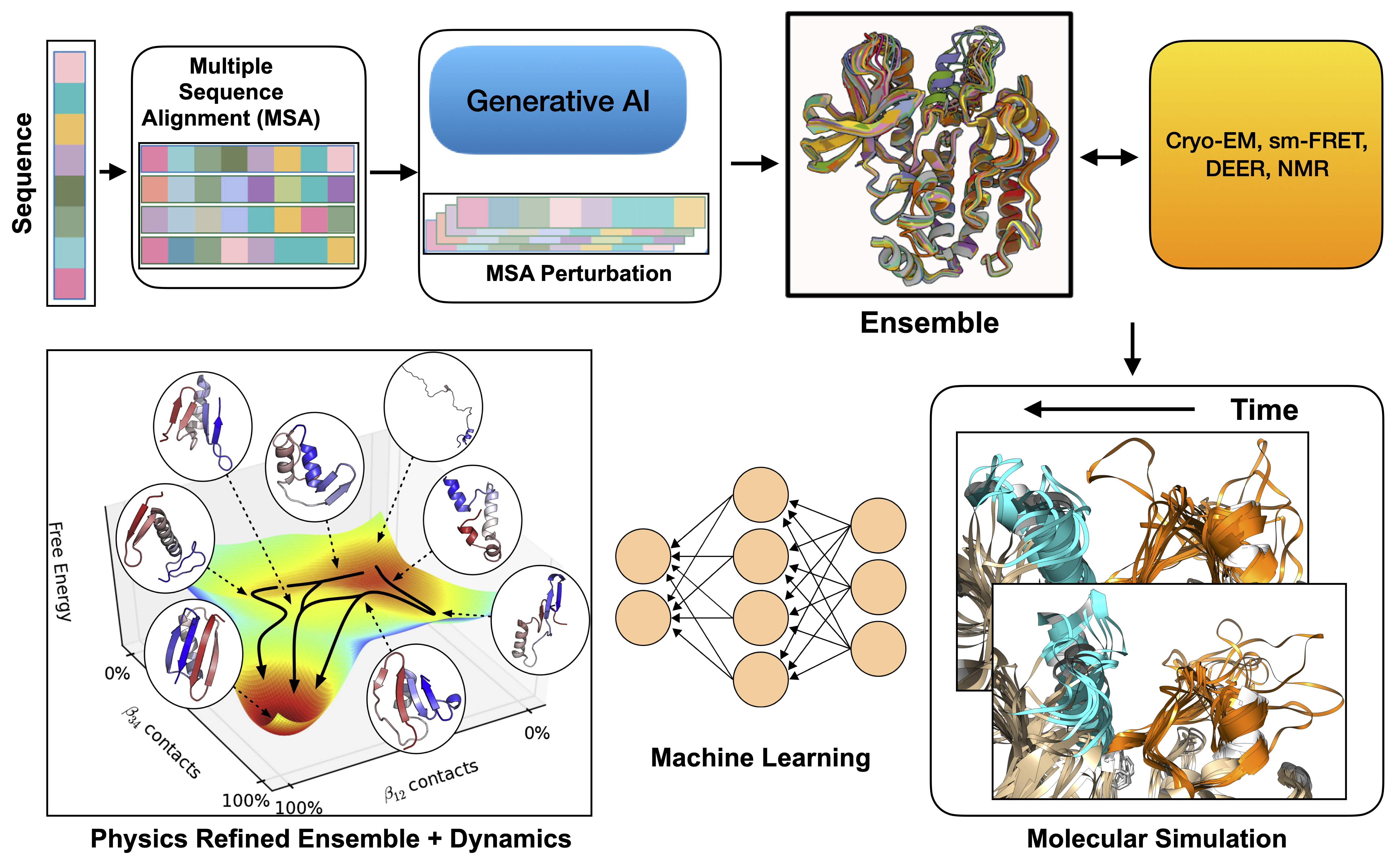
Fusing generative AI, statistical physics and experiment to capture biomolecular dynamics
Proteins consist of amino acids that can fold into multiple different structures, and depending on their structures, they can modulate diverse biological pathways within our cells. The primary dogma of computational structural biology has been to predict the native structure of proteins from their sequence, encompassing a “sequence → structure → function” paradigm. AlphaFold and other generative AI methods enabled accurate prediction of single protein structure from sequence . However it does not capture how proteins evolve over time. Environmental triggers such as temperature, light, disease causing mutations, or drug binding can shift both the populations and the transition times between these interchangeable conformations. My research aims to integrate generative AI based ensemble prediction methods with statistical physics to understand biomolecules in motion and enable “sequence → dynamics → function” paradigm. A sub interest of my research also aims to combine single molecule experiments such as cryo-EM, sm-FRET, DEER, NMR with generative AI and statistical physics to build experimental aware dynamics models of biomolecules. I see a future where the physics refined ensemble for specific protein classes can answer key biological questions such as how to find hidden druggable pockets, can we predict effect of mutations on protein dynamics and small molecule binding, can we predict asymmetry in the protein-protein interface, the questions which moves drug discovery forward.
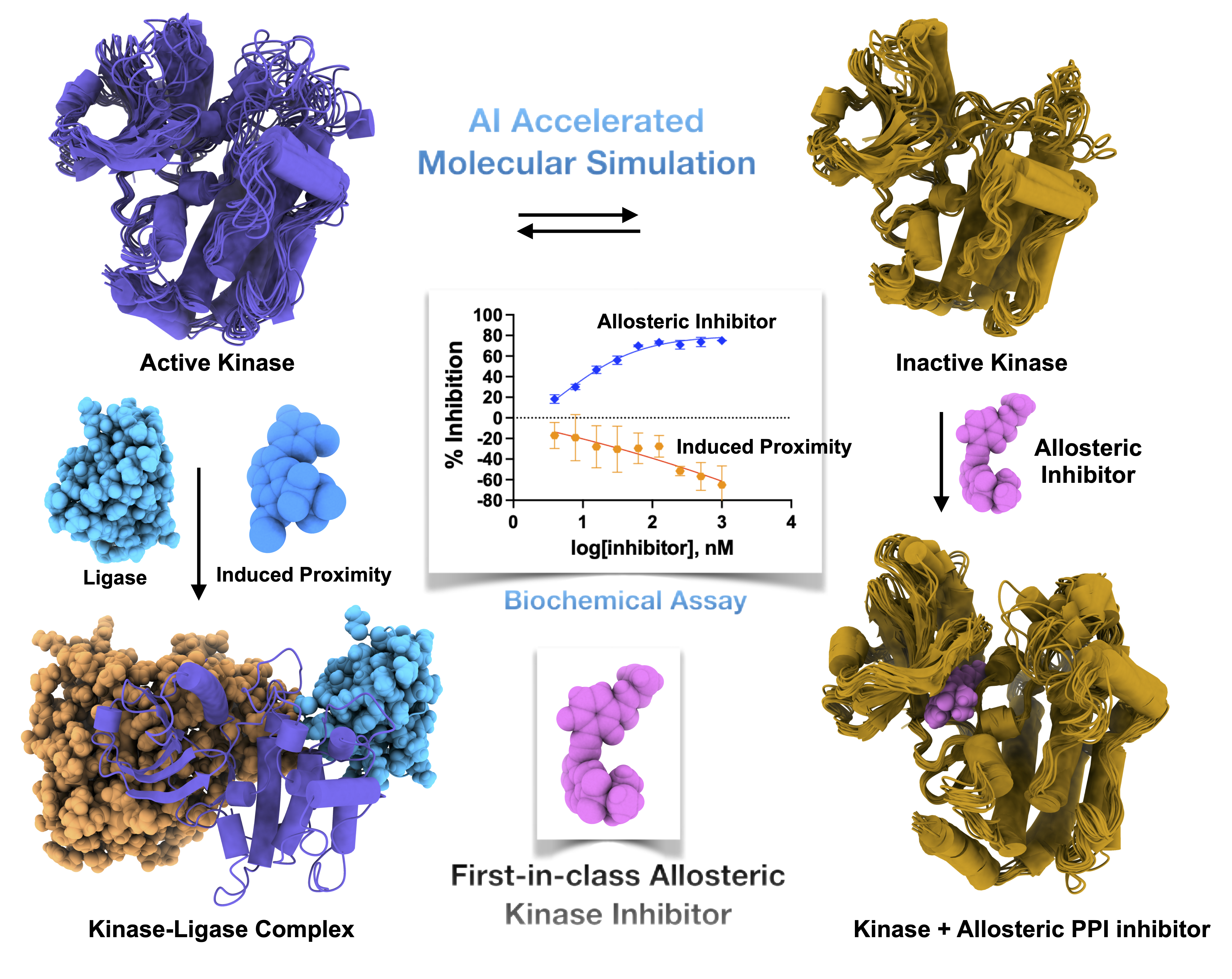
Develop allosteric modulators governing disease signalling
Protein–protein interactions regulate signaling pathways across a broad spectrum of human diseases, including inflammation, cancer, neurodegeneration, and infectious diseases. My research addresses how conformational dynamics within one protein partner can either enhance or inhibit its interaction with another, thereby modulating downstream signaling. This work raises a fundamental question: can we rationally design small molecules that stabilize or disrupt specific protein–protein interactions to control signaling outcomes?
To address this, I have partnered with experimental research groups to build an integrated theoretical and experimental framework that examines how ligand binding differentially modulates protein–protein interactions. Because such interactions are central to kinases, GPCRs, and other protein families, I aim to develop protein class specific yet generalizable models of allostery. These models seek to explain how ligand binding or mutations reshape conformational ensembles and, in turn, alter signaling pathways. I believe this approach will enable the development of a new class of therapeutics targeting protein–protein interactions across a wide range of human diseases.
Protein design meets dynamics
I am interested in exploring how we can design state specific de novo mini protein binders and antibodies that stabilize functionally important allosteric states across protein classes. This concept uses de novo binders or antibodies as probes to selectively stabilize allosteric states that are otherwise difficult to resolve using experimental techniques such as cryo-EM or X-ray crystallography. This approach has the potential to become a transformative platform for revealing functionally relevant alternate states, advancing our understanding of biological mechanisms, and expanding the scope of therapeutic modalities.
