Aromatic side-chain flipping acts a probe to understand conformational dynamics of biomolecules. In this post, we will see how to predict transition kinetics associated with different side-chain rotameric conformations using Markov State Modelling (MSM). MSM allows one to predict kinetics and equilibrium population associated with conformational dynamics. In this post we will use BACE1 as a model system. BACE1 is a target for Alzheimer’s disease. Conserved tyrosine present in flap of BACE1 governs conformational dynamics as highlighted by Bhakat and Soderhjelm (Bhakat, S.; Soderhjelm, P. Flap Dynamics in Pepsin-Like Aspartic Proteases: A Computational Perspective Using Plasmepsin-II and BACE-1 as Model Systems. Journal of Chemical Information and Modeling 2022, 62, 914-926).
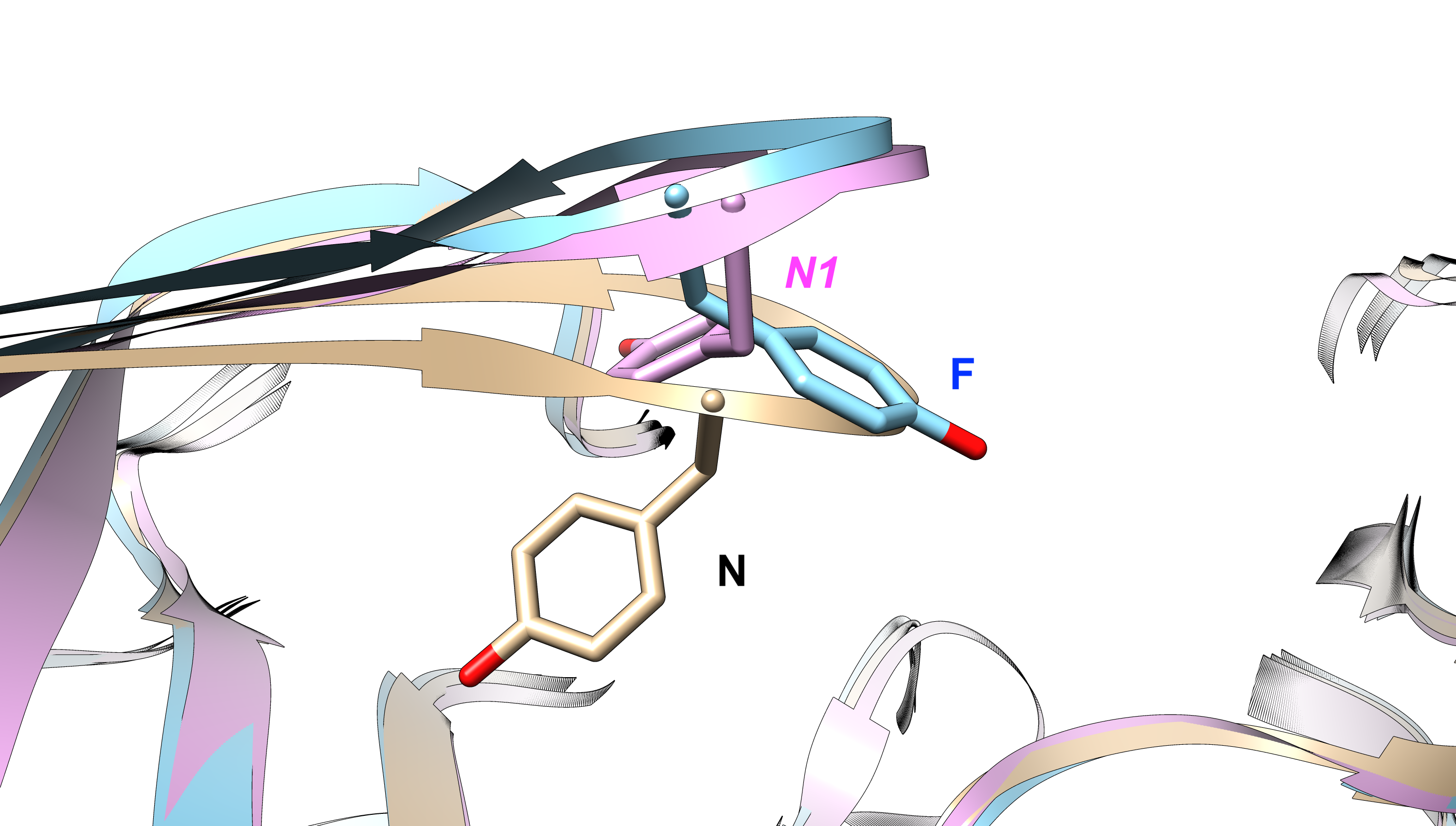
We have generated ~16 microsecond molecular dynamics simulation to sample N to F transition and vice-versa.
In order to sample transition we will first featurize our molecular dynamics data onto sin/cos transformed χ1 and χ2 angles of Tyrosine and use the features to do clustering and generate MSM. The following script does just that.
Next we will use features.pkl to do clustering using micro state K-means algorithm with K=500 (see the notebook here: https://github.com/sbhakat/MSM-ring-flipping/blob/main/pcca-msm.ipynb)
We will then perform an implied timescale test. We can clearly see from Figure 2 that the a lag time 200 is a good choice for MSM.

MSM with lag time 200 allowed us to predict equilibrium weighted free energy surface along χ1 and χ2 angles of Tyrosine (Figure 3).
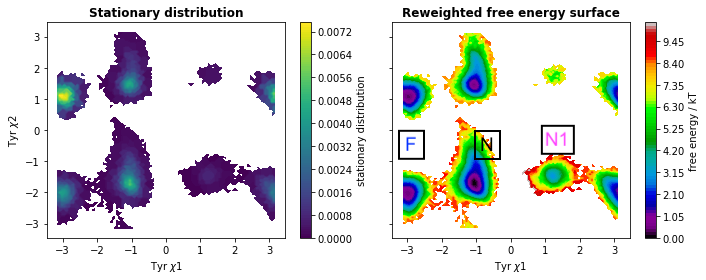
Finally we will generate coarse grained representation of MSM using PCCA. PCCA managed to separate different rotational states of Tyr as shown in Figure 4.

Finally we will calculate kinetics of ring flipping using msm.mfpt() module of PyEMMA.

Here I show how to predict kinetics associated with ring flipping which is fast flipping event (< 1 ms). One can use NMR 13C relaxation dispersion experiment to validate our MSM predicted values.
